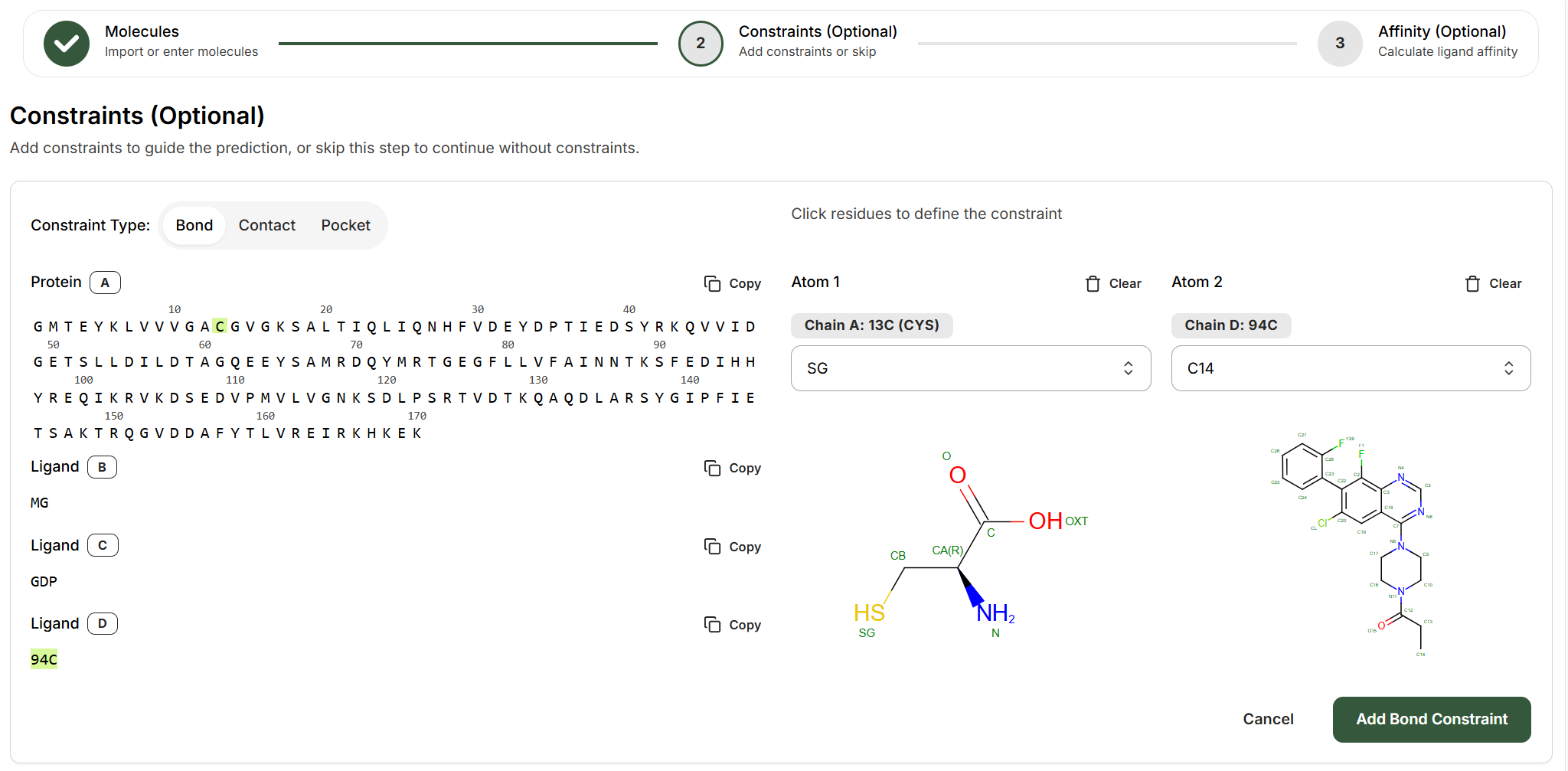
Bond Constraints
Bond constraints define covalent bonds between specific atoms that are essential for modeling covalent attachments such as irreversible inhibitors (e.g., EGFR covalent drugs), peptide cyclization, or disulfide bonds.How to Set Up a Bond Constraint
Select the two entities that will form the bond
Choose any two entities that will form the bond (protein chains, ligands, DNA, RNA) from the left panel. For example:
- Protein A and Ligand D for a covalent inhibitor
- Protein A and Protein A for a disulfide bond between residues
- Protein A and DNA B for a covalent DNA-protein crosslink
Identify the bonding atoms
On the right side, use the dropdown menus to specify which atoms will form the covalent bond:Atom 1 (Protein side):
- Chain: The protein chain letter (e.g., Chain A)
- Residue: The residue number and identity (e.g., 13C CYS)
- Atom: Select from available atoms (e.g., SG for cysteine sulfur)
- Chain: The ligand chain letter (e.g., Chain D)
- Residue: Ligand identifier (e.g., 94C)
- Atom: Select the reactive atom (e.g., C14 for acrylamide carbon)
Contact Constraints
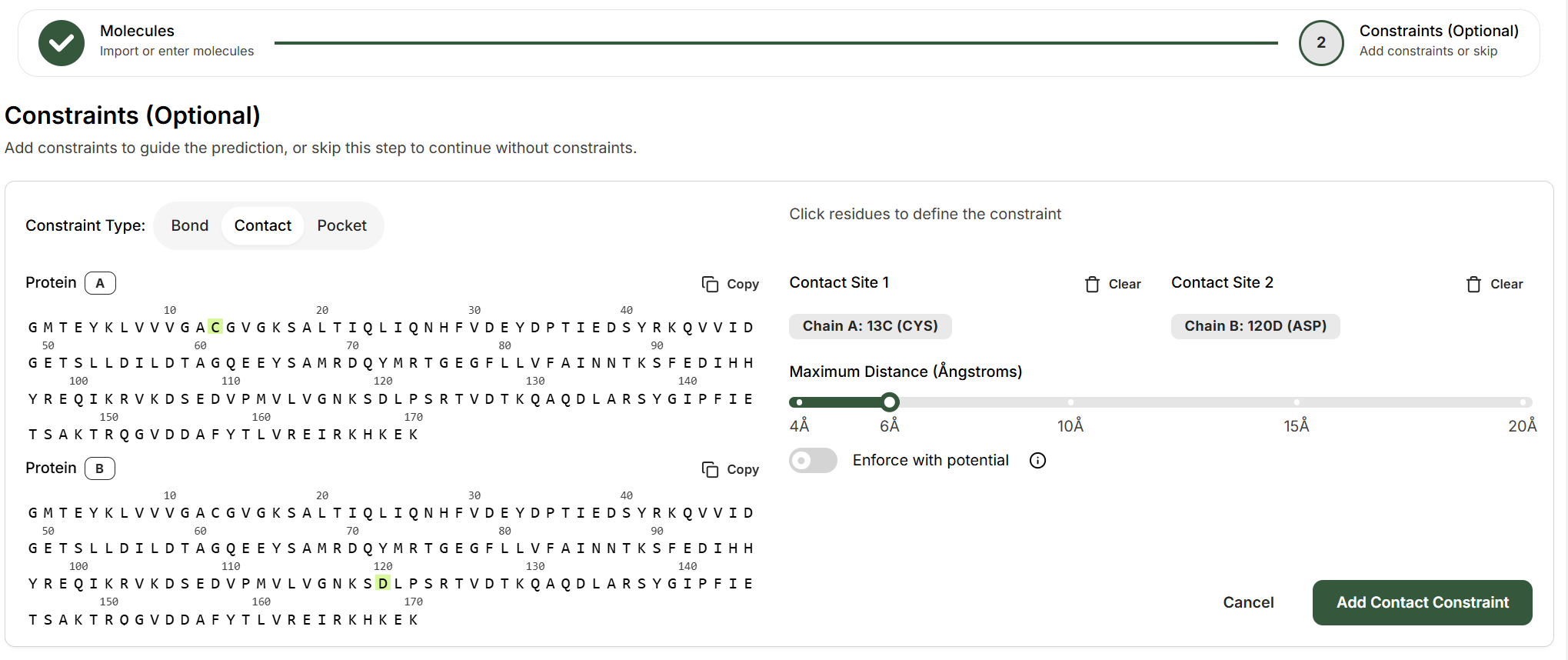
Contact constraints define distance restraints between atoms or residues, ideal for known interaction sites.How to Set Up a Contact Constraint
Select the two entities for the contact
Choose any two entities that should be in proximity. Contacts can be between any pair of biomolecules (protein, ligand, DNA, RNA), for example:
- Protein-protein: Two different residues
- Protein-ligand: A protein residue and a ligand
Specify atoms (if ligand is involved)
Depending on the entities selected, you may need to specify which atoms participate in the contact. For small molecules (ligands), select the specific atom. For polymer residues (protein, DNA, RNA), the constraint applies to one atom in the residue.
Set the maximum distance
Use the slider to define the maximum allowed distance between the contact sites, ranging from 4Å to 20Å:
- 4-6Å: Tight interaction (hydrogen bonds, salt bridges)
- 8-12Å: General proximity (nearby residues)
- 15-20Å: Loose guidance (same binding region)
Optional: Enforce with potential
Toggle “Enforce with potential” to make the distance constraint stronger. This leverages the Boltz-steering technique to enforce the distance constraint.
- Molecular glues or bridging molecules requiring proximity between biomolecular chains
- Enforcing a known H-bond or salt bridge interaction
- Directing a binder toward a specific protein region without forcing exact geometry
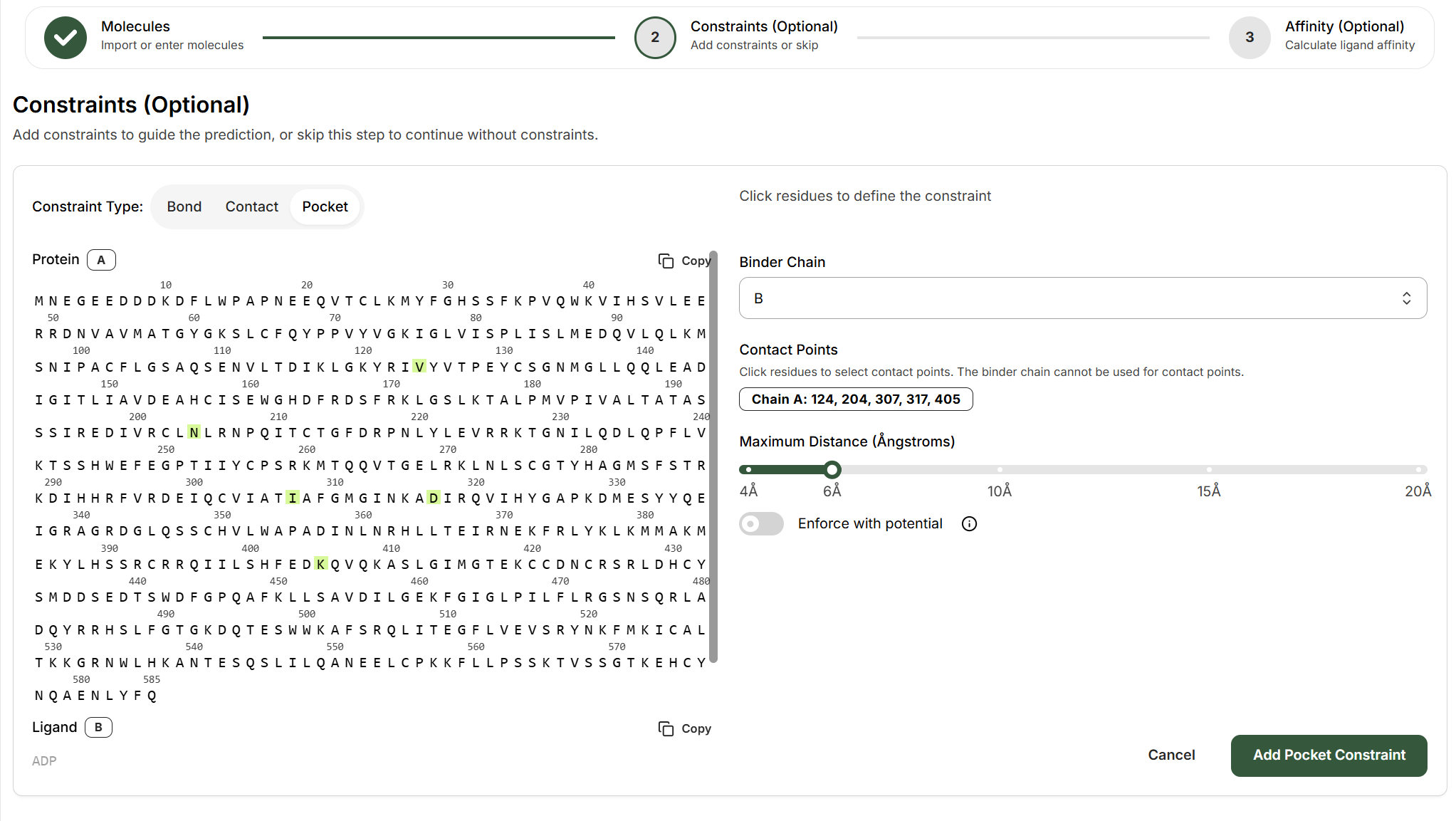
Pocket Constraints
Pocket constraints direct a binder to a specific region of the target protein by defining which residues form the binding site. This can be used for example to specify ligands binding to cryptic pockets, or allosteric sites, or specifying the epitope that an antibody should bind to on the antigen.How to Set Up a Pocket Constraint
Select the binder chain
From the Binder Chain dropdown, choose which entity should bind to the defined pocket (e.g., Chain B).
Define the binding pocket
Click on residues in the protein sequence viewer to select the residues that form your target binding site. Selected residues will be highlighted in green.The interface displays: Chain A: 124, 204, 307, 317, 405 (example residues)
Set the maximum distance
Use the slider to define how close the binder must be to the pocket residues (4-20Å). This sets the “binding site radius.”
Optional: Enforce with potential
Toggle “Enforce with potential” to strengthen the pocket constraint. The model force itself to place the binder near the specified residues within the specified distance.
- Targeting an allosteric site distinct from the orthosteric pocket
- Modeling binders to a cryptic pocket that’s not obvious in the apo structure
- Disambiguating between multiple potential binding sites on the same protein
- Enforcing binding to a specific sub-pocket in a large binding cavity
Tips for Effective Constraints
Start Simple
Add one constraint at a time and validate results before adding more. Over-constraining can force unrealistic structures.
Use Structural Evidence
Base constraints on SAR data, crystal structures, or biochemical assays.
Test Without Constraints First
Run a prediction without constraints as a baseline. If it fails to find the correct binding mode, then add constraints.
Validate Confidence Scores
Check pLDDT and iPTM scores after adding constraints. Lower scores may indicate incompatible constraints.
What’s Next?
Bond Example: Covalent Inhibitors
Walk through a real covalent warhead prediction
Contact Example: Molecular Glues
Model ternary complexes with contact constraints
Pocket Example: Cryptic Sites
Target allosteric or cryptic binding pockets